
—— Samarbeidsstudie av Zhejiang CDC, Macro & Micro-Test og China CDC Publisert i Frontiers in Cellular and Infection Microbiology
Studieoversikt
I mai 2026 publiserte Frontiers in Cellular and Infection Microbiology (JCR Q1, IF ≈ 4.6) en artikkel ledet av Zhejiang Provincial Center for Disease Control and Prevention (Zhejiang CDC), med bioinformatikkteamet fra Beijing Macro & Micro-Test Bio-Tech Co., Ltd. og National Institute for Communicable Disease Control and Prevention (China CDC) som medforfattere. Studien har tittelen:
"Identifisering og fylogenetisk analyse av syv Brucella abortus-stammer i Zhejiang, Kina."

Denne studien representerer den første systematiske, helgenombaserte fylogenetiske sporbarhetsanalysen av Brucella abortus (B. abortus) i Zhejiang-provinsen i Kina. Teamet analyserte sju isolater samlet inn fra 2015 til 2025 (fire stammer av menneskelig opprinnelse og tre stammer av bovin opprinnelse fra Jinhua, Quzhou og Ningbo). Funnene gir genomiske bevis for opprinnelsen og overføringsveiene til denne «nordlige dominerende arten» i en atypisk sørlig epidemisk region i Øst-Kina.
Bakgrunn og betydning
Brucellose er en zoonotisk sykdom forårsaket av bakterier av slekten Brucella. Brucella abortus infiserer primært storfe, men kan også forårsake sykdom hos mennesker. I Kina viser brucellose markert geografisk variasjon: den høyeste forekomsten forekommer i nordlige provinser (f.eks. Indre Mongolia, Shanxi, Heilongjiang). Derimot har sørlige provinser, inkludert Zhejiang, historisk sett vært dominert av Brucella melitensis, med svært få rapporterte tilfeller av B. abortus. Denne regionale forskjellen gjør genetisk karakterisering og kildesporing av B. abortus i Zhejiang til en viktig folkehelseprioritet.
Metoder og viktige funn
Forskningsteamet tok i bruk en flerstrenget strategi som kombinerer molekylærbiologi og bioinformatikk:
1.Patogenidentifikasjon og grunnleggende typing
BCSP-31-gen-PCR og AMOS-PCR bekreftet at alle syv isolatene var B. abortus.
Multilokussekvenstyping (MLST) basert på ni husholdningsgener avslørte at alle isolatene tilhørte sekvenstype ST2, noe som indikerer høy genetisk homogenitet blant de sirkulerende B. abortus-stammene i Zhejiang.
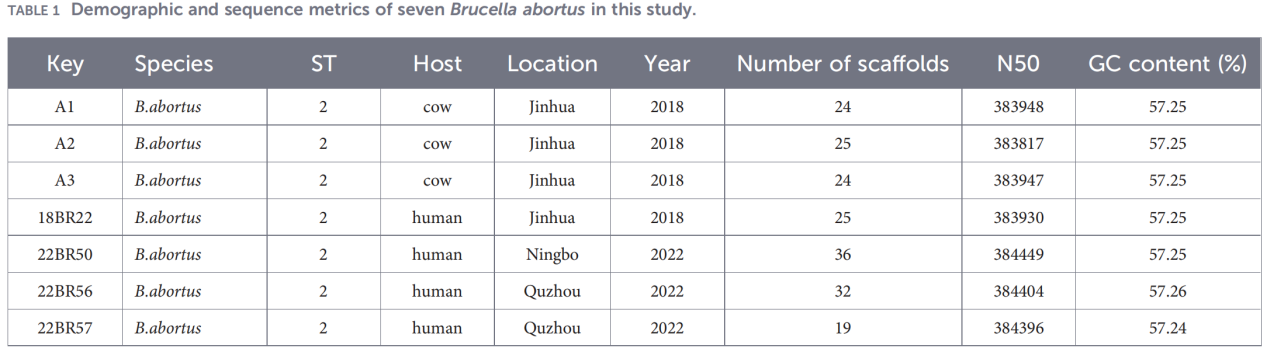
2.Helgenomkarakterisering
Helgenomsekvensering ble utført på Illumina NovaSeq-plattformen. Analyse av gjennomsnittlig nukleotididentitet (ANI) viste at Zhejiang-isolatene hadde opptil 99,99 % likhet med referansestammen B. abortus 544.
Pan-genomanalyse avdekket en svært konservert populasjon: 3084 kjernegener ble identifisert, sammen med bare 10 skallgener, og ingen myke kjerne- eller skygener ble oppdaget.
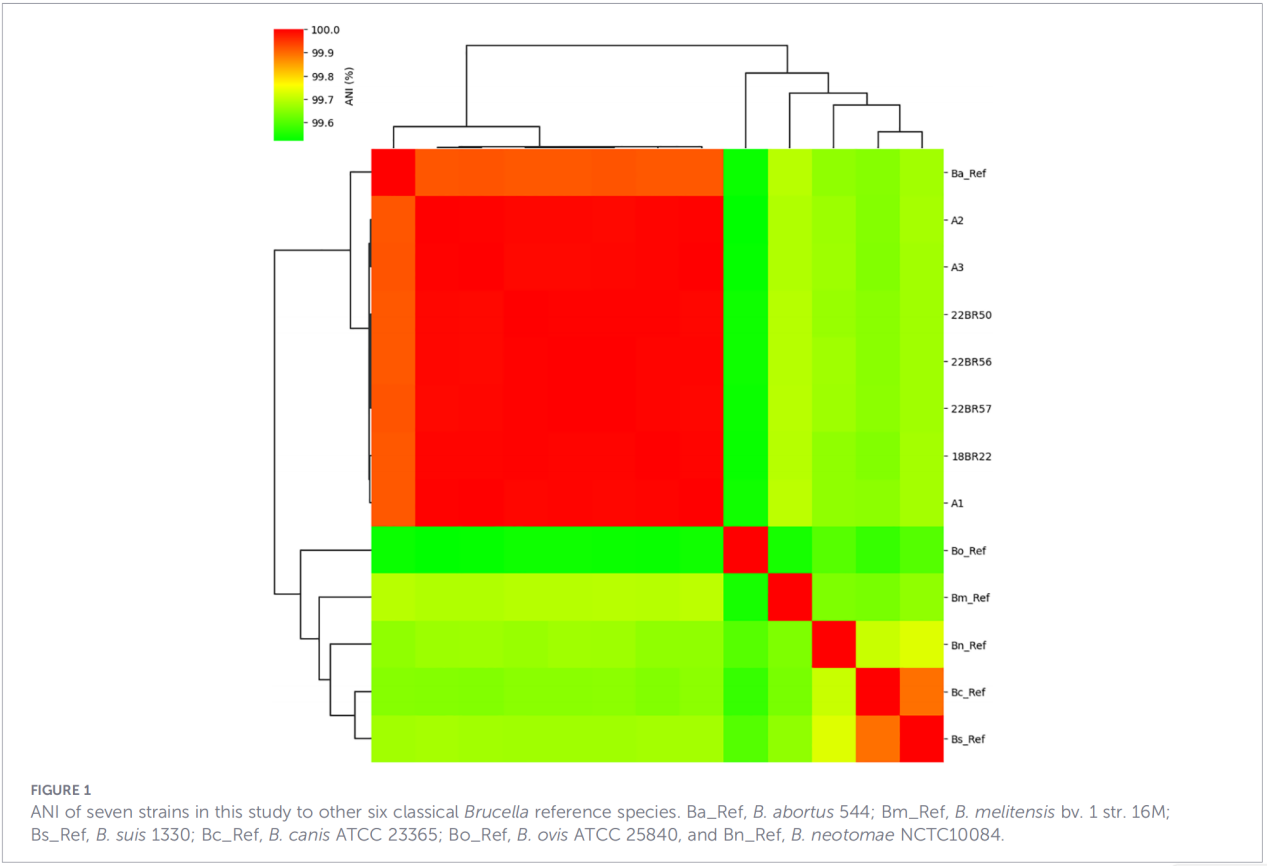
3.Genprofiler for virulens og antimikrobiell resistens
Totalt 68 virulensrelaterte faktorer ble predikert, som dekket klassiske veier som LPS-biosyntese, T4SS-sekresjonssystemet og BvrR-BvrS tokomponents reguleringssystem. Det er verdt å merke seg at alle isolatene manglet adhesin-genene bmaA og btaF. Resistensgenanalyse oppdaget kun mprF-genet i CARD-databasen, uten andre resistensdeterminanter identifisert.
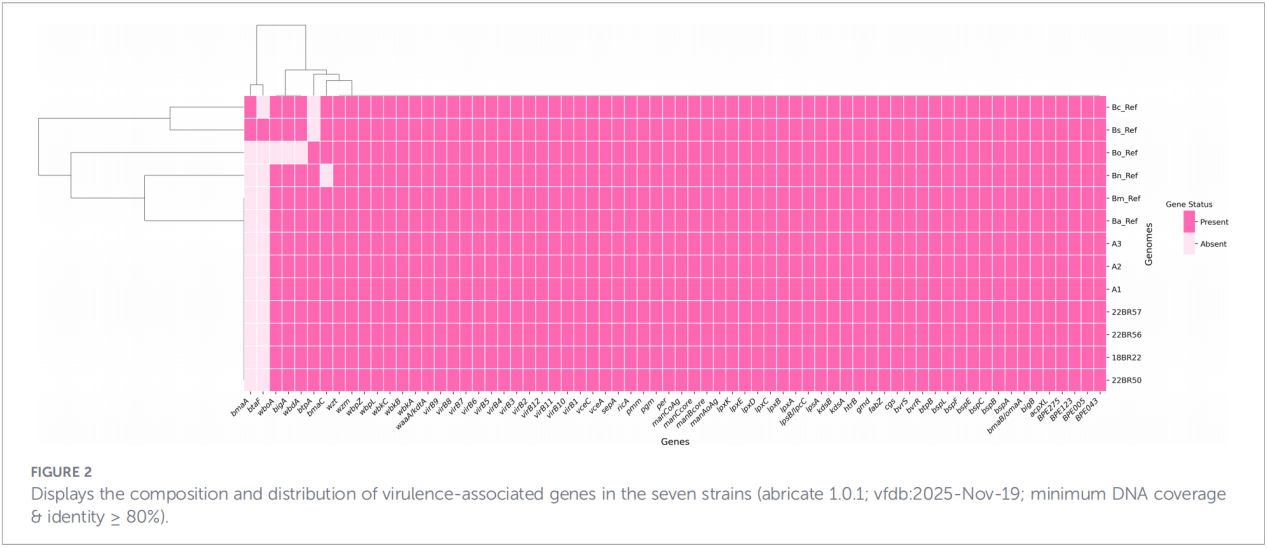 4. Fylogenetisk rekonstruksjon og overføringssporing
4. Fylogenetisk rekonstruksjon og overføringssporing
Kjernegenomanalyse av enkeltnukleotidpolymorfisme (cgSNP) plasserte Zhejiang-isolatene på en spesifikk posisjon i det globale fylogenetiske treet. Resultatene viste at Zhejiang-stammene danner en monofyletisk gruppe sammen med stammer fra Russland, Mongolia og flere nordlige kinesiske provinser (Ningxia, Heilongjiang, Indre Mongolia, Hebei, Gansu, Beijing). Denne gruppen deler seg videre inn i tre distinkte underklader (klade 1–3), noe som tyder på flere uavhengige introduksjonshendelser.
Konklusjoner og implikasjoner
Denne studien gir det første svært presise genomiske datasettet for B. abortus i Zhejiang-provinsen og gir flere viktige konklusjoner:
- Clear genetisk bakgrunn– B. abortus-stammene som sirkulerer i Zhejiang tilhører ST2, er genomisk svært konserverte og representerer en typisk bovin brucellose-avstamning.
2. Evidensiteten til kryssregional overføring– Fylogenetisk analyse støtter ikke eksistensen av en uavhengig endemisk avstamning i Zhejiang. Dataene tyder i stedet sterkt på at disse stammene stammer fra Nord-Kina og kan dele en felles evolusjonær bakgrunn med stammer fra Russland og Mongolia. Tilstedeværelsen av tre underklader impliserer flere separate introduksjonshendelser.
3. Folkehelsemessige implikasjoner– Funnene understreker verdien av genomisk overvåking av brucellose, selv i tradisjonelt ikke-endemiske regioner som Zhejiang. Selv om det nåværende tilfelletallet er lavt, kan høyoppløselige verktøy som cgSNP effektivt spore kilden til importerte utbrudd og gi vitenskapelig bevis for å avbryte smittekjeder knyttet til transport av husdyr mellom provinsielle regioner.
Dette arbeidet fyller ikke bare et forskningsgap i Zhejiang-provinsen, men gir også nye basisdata for patogenovervåking og risikovurdering av brucellose i Yangtze-elvedelta-regionen.
Papirinformasjon:
Yang, Y., Shi, X., Chen, J., Wang, L., Wu, Z., Yao, W., … & Wu, B. (2026). Identifisering og fylogenetisk analyse av sju Brucella abortus-stammer i Zhejiang, Kina. Frontiers in Cellular and Infection Microbiology, 16, 1758965.
Publisert: 10. juni 2026